Research
Our goal is to understand the relationship between protein structure and function. In particular, we are focused on the interaction between proteins and their ligands (small-molecule ligands) and how the interaction triggers protein activation/inhibition.
Experimental Structural Biology
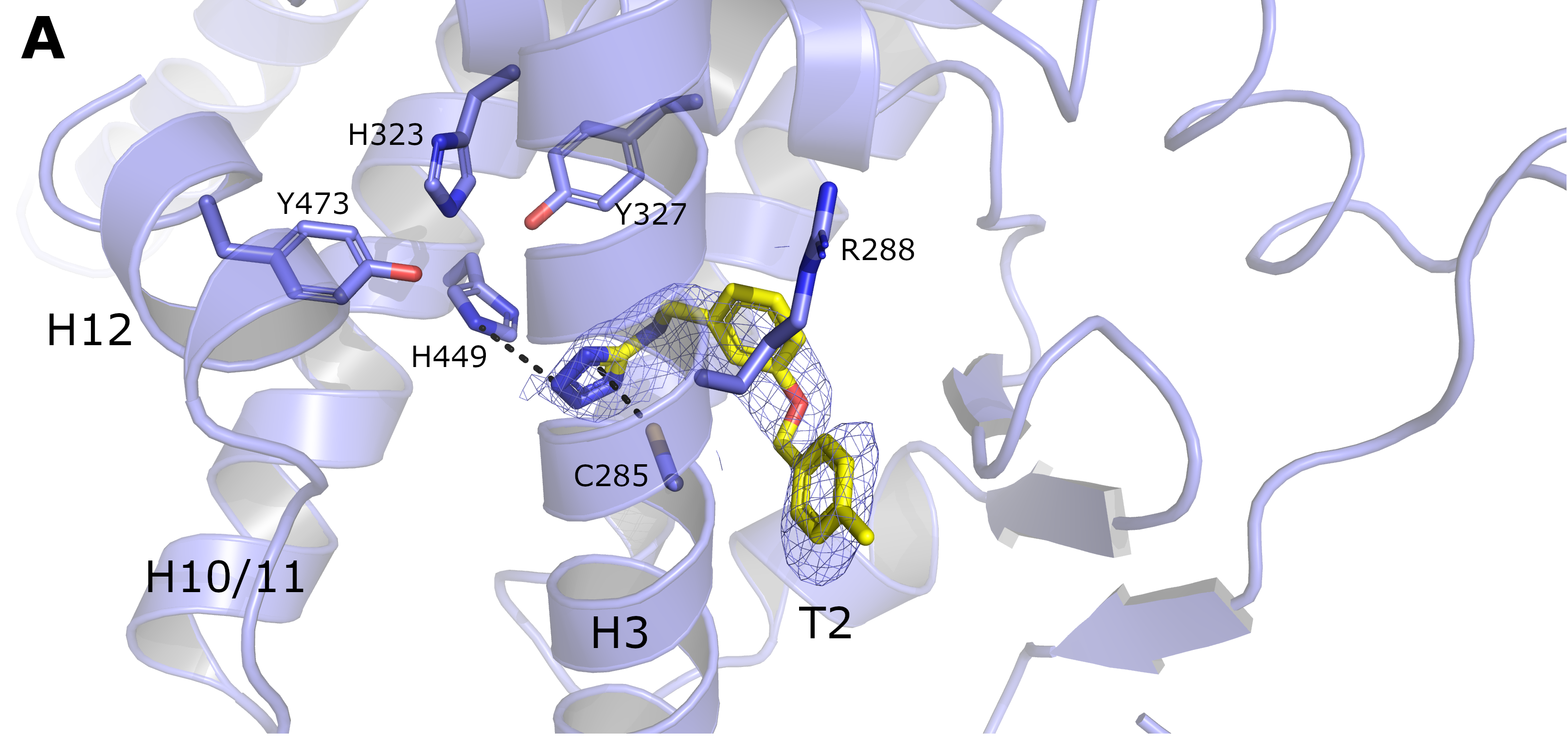
Our group is very interested in advancing the structural knowledge of proteins that could serve as potential targets for bacterial antibiotics. The increasing observed rates of bacterial resistance to antibiotics is quite concerning. It becomes even worse by the limited number of pathways explored by the existing antibiotics. Our approach to tackle this problem is based on the proposition of new antibiotic targets, together with their structural determination and the structure-based proof of concept. One recent example of this branch of our research was recently published in Biochimie. More interesting results coming soon!
Computational Structural Biology
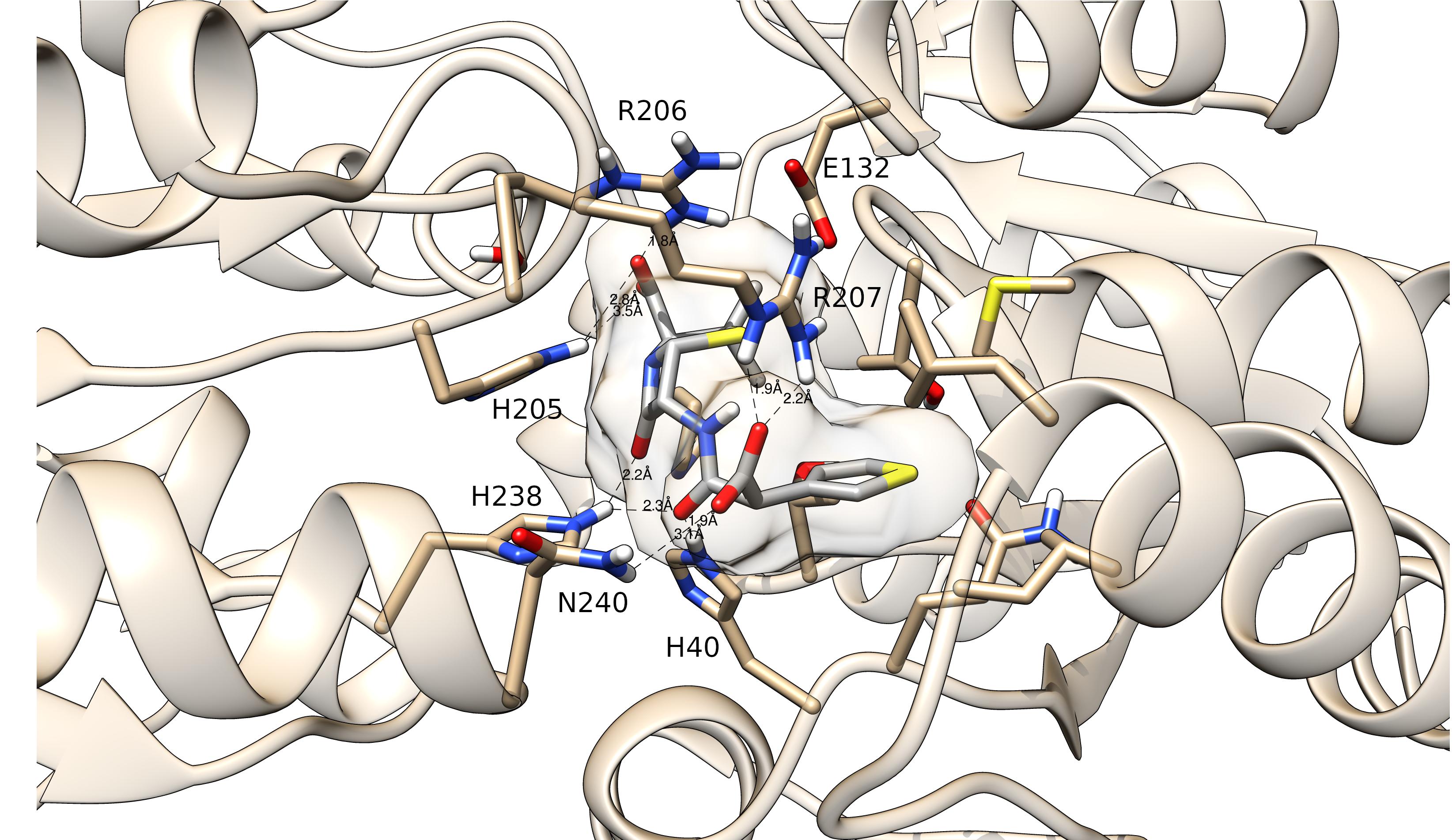
Moving forward, we also believe that we can get a better understanding of the structure-function relationship by modeling. In this context, our group has also dedicated some efforts in the modeling of protein-ligand complexes in a structure-based approach. In this branch of our research, we have been developing tools such as MolShaCs, and LiBELa.
MolShaCs is a tool dedicate to screen ligands using a ligand-based similarity with 3D descriptors. In MolShaCS, the descriptors are based on the molecular volume and charge distribution, as previously described.
LiBELa is a ligand docking engine that is based on the ligand similarity as well as by the ligand-receptor interactions. In the first stage of the docking, the ligand is pre-docked in the protein binding pocket by maximizing the overlap of the MolShaCs 3D descriptors in the Cartesian space between the search ligand and reference ligand bound to the target protein. In the second stage, the pre-docked ligand is optimized in the Cartesian space by minimizing the interaction energy. The interaction energy accounts for VDW and polar terms, as well as hydrogen bonds and ligand- and receptor desolvation. LiBELa has also been recently adapted to allow Monte Carlo (MC) simulations of the ligand within the protein binding pocket.
